
Hóa sinh học arsenic

Hóa sinh học arsenic là thuật ngữ để nói tới các quá trình hóa sinh học có sử dụng arsenic hoặc các hợp chất của nó, chẳng hạn các arsenat. Arsenic là nguyên tố hóa học khá phổ biến trong lớp vỏ Trái Đất (~ 1,8-2,1 ppm), và mặc dù nhiều hợp chất của arsenic thường được coi là có độc tính cao đối với phần lớn các dạng sự sống, nhưng một loạt các hợp chất arsenic hữu cơ được sản sinh theo con đường sinh học, với nhiều dạng sinh vật có hoạt động trao đổi chất một loạt các hợp chất hữu cơ và vô cơ của arsenic. Mô hình này là chung cho các nguyên tố có liên quan khác, chẳng hạn như selen, những nguyên tố có thể thể hiện cả các tác động có lợi lẫn có hại. Hóa sinh học arsenic đã trở thành vấn đề có tính thời sự do nhiều hợp chất arsenic độc hại được tìm thấy trong một số tầng ngậm nước,[1] có tiềm năng ảnh hưởng tới nhiều triệu người thông qua các quá trình hóa sinh học.[2]
Nguồn cung cấp arsenic
[sửa | sửa mã nguồn]Các hợp chất arsenic hữu cơ trong tự nhiên
[sửa | sửa mã nguồn]
Chứng cứ cho rằng arsenic có thể là chất dinh dưỡng có lợi khi ở mức dấu vết dưới ngưỡng mà các sinh vật thường phơi nhiễm đã từng được xem xét lại.[3] Một số hợp chất arsenic hữu cơ tìm thấy trong tự nhiên là arsenobetain và arsenocholin,[4] với cả hai đều được tìm thấy trong nhiều loài sinh vật biển.[2] Một số nucleoside chứa As (các dẫn xuất đường) cũng được biết đến.[5] Một số các hợp chất arsenic hữu cơ sinh ra từ các quá trình methyl hóa. Chẳng hạn, loài nấm mốc Scopulariopsis brevicaulis sinh ra một lượng đáng kể trimethylarsin nếu có mặt các hợp chất arsenic vô cơ.[6] Hợp chất arsenic hữu cơ arsenobetain được tìm thấy trong một số thực phẩm nguồn gốc từ biển như cá và tảo cũng như trong một số loài nấm với hàm lượng tương đối lớn. Mức hấp thụ arsenic trung bình của con người là khoảng 10–50 µg/ngày. Các giá trị ở mức khoảng 1000 µg là không bất thường sau khi ăn cá hoặc nấm; tuy nhiên, có rất ít nguy hiểm trong việc ăn cá do hợp chất này của arsenic là gần như vô hại.[7]
- Các hợp chất arsenic hữu cơ đại diện được tìm thấy trong tự nhiên.
-
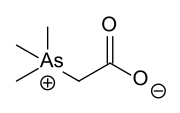 Arsenobetain, một trong các hợp chất arsenic hữu cơ phổ biến nhất trong tự nhiên. Cũng phổ biến còn có arsenocholin, với nhóm CH2OH thế chỗ cho nhóm CO2H).
Arsenobetain, một trong các hợp chất arsenic hữu cơ phổ biến nhất trong tự nhiên. Cũng phổ biến còn có arsenocholin, với nhóm CH2OH thế chỗ cho nhóm CO2H). -
 Trimethylasin, được sinh ra từ phản ứng vi sinh đối với các sắc tố chứa arsenat.
Trimethylasin, được sinh ra từ phản ứng vi sinh đối với các sắc tố chứa arsenat. -
 Dẫn xuất ribose chứa arsenic (R = một vài nhóm)
Dẫn xuất ribose chứa arsenic (R = một vài nhóm)



Nguồn cục bộ chứa arsenic là các chất nhuộm màu xanh lục, một thời phổ biến trong các loại giấy dán tường, như lục Paris. Một loạt các loại bệnh tật đã từng được quy cho hợp chất này, mặc dù độc tính của nó đã bị thổi phồng.[8]
Trimethylasin ((CH3)3As), một thời được biết đến như là khí Gosio, là một hợp chất arsenic hữu cơ nặng mùi được sinh ra khá phổ biến trong các phản ứng vi sinh trên các chất nền chứa arsenic vô cơ.[9]
Các hợp chất arsenic hóa trị 5 dễ dàng bị khử thành hợp chất arsenic hóa trị 3 và có thể đã từng có vai trò như là một tác nhân nhận electron trên Trái Đất nguyên thủy.[10] Các hồ chứa một lượng đáng kể arsenic vô cơ hòa tan là nơi nuôi dưỡng các vùng sinh vật chịu được arsenic.
Các tuyên bố không chính xác về sự sống dựa trên arsenic
[sửa | sửa mã nguồn]Mặc dù các phosphat và arsenat là tương tự về cấu trúc, nhưng không có chứng cứ cho thấy là arsenic thay thế cho phosphor trong DNA hay RNA.[11][12][13] Một thực nghiệm năm 2010 sử dụng vi khuẩn GFAJ-1 để ra tuyên bố về sự thay thế này đã bị phủ nhận vào năm 2012.
Các hợp chất arsenic nguồn gốc nhân tạo
[sửa | sửa mã nguồn]Các nguồn arsenic nhân tạo (do con người tạo ra), giống như các nguồn tự nhiên, chủ yếu là các oxide arsenic cùng các anion có liên quan. Các nguồn arsenic nhân tạo, bao gồm cả phế thải từ khai thác chế biến khoáng sản, các trang trại chăn nuôi lợn và gia cầm.[14] Chẳng hạn, nhiều loại quặng, đặc biệt là các loại khoáng vật sulfide, đều chứa arsenic ở một hàm lượng nào đó và nó được giải phóng ra trong quá trình thiêu kết (đốt trong không khí). Trong các quá trình như vậy, arsenide được chuyển hóa thành arsenic trioxide (As2O3), là chất bay hơi ở nhiệt độ cao (> 465 °C) và được giải phóng vào khí quyển. Các trang trại chăn nuôi gia cầm và lợn cũng từng sử dụng nhiều hợp chất arsenic hữu cơ roxarsone như là một loại kháng sinh trong thức ăn.[15][16] Một số loại gỗ được xử lý bằng đồng arsenat trong vai trò của chất bảo quản. Cơ chế mà theo đó các nguồn này tác động tới các sinh vật trong chuỗi kế tiếp là chưa rõ nhưng có lẽ là đa dạng. Một trong những cách thức được nhắc tới nhiều nhất là methyl hóa.[17]
Acid monomethyl hóa, acid methanearsonic (CH3AsO(OH)2), là tiền chất cho một số loại thuốc diệt nấm (tên thương mại Neoasozin) trong gieo trồng lúa và bông. Các dẫn xuất của acid phenylarsonic (C6H5AsO(OH)2) được sử dụng như là phụ gia trong thức ăn chăn nuôi, bao gồm acid 4-hydroxy-3-nitrobenzenearsonic (3-NHPAA hay Roxarsone), acid ureidophenylarsonic và acid p-arsanilic. Các ứng dụng này gây nhiều mâu thuẫn do chúng đưa các dạng hòa tan của arsenic vào môi trường.
Dược phẩm chứa arsenic
[sửa | sửa mã nguồn]Mặc cho, hoặc có lẽ là do, độc tính được biết đến từ lâu của nó, các loại dược phẩm và thuốc độc chứa arsenic có lịch sử trong y học và lang băm vẫn còn tiếp diễn trong thế kỷ 21.[18][19] Từ đầu thế kỷ 19 và tiếp diễn trong thế kỷ 20, dung dịch Fowler, một loại thuốc độc pha chế từ kali arsenit (KAsO2), đã từng được mua bán như một loại thuốc để điều trị ung thư bạch cầu. Hợp chất arsenic hữu cơ Salvarsan (arsphenamine, hợp chất 606) là tác nhân hóa học trị liệu tổng hợp đầu tiên để điều trị giang mai và bệnh trùng mũi khoan, do Paul Ehrlich phát minh.[19] Tuy nhiên, việc điều trị này dẫn tới nhiều vấn đề gây ra các biến chứng sức khỏe kéo dài.[20] Vào khoảng năm 1943 nó cuối cùng đã bị thay thế bằng penicillin. Loại thuốc có liên quan là Melarsoprol vẫn còn được sử dụng để điều trị bệnh trùng mũi khoan (bệnh ngủ Trypanosoma châu Phi) giai đoạn cuối, mặc dù độc tính cao và các tác dụng phụ có thể gây tử vong của nó.
Các nghiên cứu in vitro gợi ý rằng arsenic trioxide (As2O3) ngăn ngừa sự phát triển của các tế bào u tủy xương thông qua ngăn chặn chu trình tế bào cũng như khởi động sự chết tế bào.[21] Các kết quả này gợi ý rằng arsenic trioxide có thể là hữu ích lâm sàng trong điều trị các bệnh nhân với bệnh u tủy xương đa phát[21] hay ung thư bạch cầu.[22]
Methyl hóa arsenic
[sửa | sửa mã nguồn]Arsenic vô cơ và các hợp chất của nó, trong quá trình tham gia vào chuỗi thức ăn, được trao đổi chất (khử độc tính) không ngừng thông qua quá trình gọi là methyl hóa.[17] Quá trình methyl hóa xảy ra với các phản ứng methyl hóa khử và oxy hóa luân phiên, nghĩa là khử arsenic hóa trị 5 thành arsenic hóa trị 3 tiếp theo là bổ sung nhóm methyl (CH3).[23]

Ở động vật có vú, methyl hóa xảy ra trong gan bởi các methyltransferase, các sản phẩm là (CH3)2AsOH (acid dimethylarsinơ) và (CH3)2As(O)OH (acid dimethylarsinic), có trạng thái oxy hóa tương ứng là As(III) và As(V).[2] Mặc dù cơ chế methyl hóa arsenic ở người vẫn chưa được sáng tỏ, nhưng nguồn methyl là methionin, và điều này gợi ý về vai trò của S-adenosyl methionin.[24] Sự phơi nhiễm trước các liều gây ngộ độc bắt đầu khi khả năng methyl hóa của gan bị vượt qua hoặc bị ngăn cản.
Có hai dạng chính của arsenic có thể thâm nhập vào cơ thể là arsenic (III) và arsenic (V).[25] Arsenic (III) đi vào các tế bào thông qua aquaporin 7 và 9, là các kiểu của aquaglyceroporin.[25] Các hợp chất arsenic (V) sử dụng các tác nhân vận chuyển phosphat để tiến vào tế bào.[25] Arsenic (V) có thể chuyển hóa thành arsenic (III) bởi enzym purin nucleoside phosphorylase.[25] Điều này được phân loại như là một bước kích hoạt sinh học, do mặc dù arsenic (III) có độc tính cao hơn, nhưng nó lại dễ bị methyl hóa hơn.[26]
Có hai lộ trình mà theo đó các hợp chất arsenic vô cơ được methyl hóa.[27]
Lộ trình thứ nhất sử dụng Cyt19 arsenic methyltransferase để methyl hóa arsenic (III) thành hợp chất arsenic (V) monomethyl hóa (đơn methyl hóa).[25] Hợp chất này sau đó được chuyển hóa thành hợp chất arsenic (III) monomethyl hóa bằng việc sử dụng Glutathion S-Transferase Omega-1 (GSTO1).[25] Hợp chất arsenic (V) monomethyl hóa sau đó cũng có thể bị methyl hóa một lần nữa bằng Cyt19 arsenic methyltransferase để tạo ra hợp chất dimethyl arsenic (V) để sau đó chuyển hóa thành hợp chất dimethyl arsenic (III) bởi Glutathion S-Transferase Omega-1 (GTSO1).[25]
Lộ trình thứ hai sử dụng glutathion (GSH) kết hợp với arsenic (III) để tạo ra phức chất arsenic (GS) 3.[25] Phức chất này có thể tạo thành phức chất arsenic (III) GS monomethyl hóa bằng sử dụng Cyt19 arsenic methyltransferase, và phức chất GS monomethyl hóa này nằm trong cân bằng với arsenic (III) monomethyl hóa.[25] Cyt19 arsenic methyltransferase có thể methyl hóa phức chất một hay nhiều lần, và nó tạo thành một phức chất arsenic GS dimethyl hóa, và nó nằm trong cân bằng với phức chất dimethyl arsenic (III).[25] Các hợp chất arsenic monomethyl hóa và dimethylat hóa đều dễ dàng bài tiết theo nước tiểu.[26] Tuy nhiên, hợp chất monomethyl hóa được chỉ ra là có hoạt tính cao hơn và độc hại hơn so với các hợp chất arsenic vô cơ đối với các hepatocyte (tế bào gan), keratinocyte ở da, và các tế bào biểu mô cuống phổi (phổi) ở người.[28]
Các nghiên cứu ở động vật thực nghiệm và người chỉ ra rằng cả arsenic vô cơ và các chất trao đổi chất methyl hóa đều đi qua nhau thai để vào bào thai. Tuy nhiên, có chứng cứ cho thấy sự methyl hóa tăng lên trong quá trình mang thai và điều này có nghĩa là nó có thể có tính bảo vệ cao hơn cho các sinh vật đang phát triển.[29]
Methyl hóa bằng enzym của arsenic là một quá trình khử độc tính; nó có thể được methyl hóa thành methylarsenit, dimethylarsenit hay trimethylarsenit, tất cả đều có hóa trị 3. Quá trình methyl hóa được xúc tác bởi arsenic methyltransferase (AS3MT) ở động vật có vú để chuyển giao một nhóm methyl trên phụ nhân tử S-adenomethionin (SAM) cho arsenic (III). Bản sao tương đồng của AS3MT được tìm thấy ở vi khuẩn và được gọi là CmArsM. Enzym này được thử nghiệm trong 3 trạng thái (không phối thể, liên kết arsenic (III) và liên kết SAM). Khu vực liên kết arsenic (III) sử dụng nhóm thiol của phần còn lại của cysein. Sự xúc tác bao gồm thiol hóa Cys72, Cys174 và Cys224. Trong phản ứng SN2, điện tích dương trên nguyên tử lưu huỳnh của SAM hút electron liên kết từ carbon của nhóm methyl, và nó tương tác với cặp electron đơn độc của arsenic để tạo ra một liên kết As−C, giải phóng S-Adenosyl-L-homocystein (SAH).[30]
Bài tiết
[sửa | sửa mã nguồn]Ở người, lộ trình chính để bài tiết phần lớn các hợp chất arsenic là thông qua nước tiểu. Chu kỳ bán thải sinh học các hợp chất arsenic vô cơ là khoảng 4 ngày, nhưng nó là hơi ngắn hơn sau khi phơi nhiễm arsenat so với phơi nhiễm arsenit. Các chất trao đổi chất chính được bài tiết trong nước tiểu của người phơi nhiễm arsenic vô cơ là các acid arsenic mono- và dimethyl hóa, cùng với một số arsenic vô cơ chưa bị trao đổi chất.[24]
Sự biến đổi sinh học của arsenic để bài tiết chủ yếu được thực hiện thông qua con đường sử dụng nhân tử hạt nhân Nrf2.[31] Trong các điều kiện bình thường, Nrf2 liên kết với protein Keap1 trong trạng thái không hoạt hóa.[32] Với sự hấp thụ arsenic vào trong tế bào và các phản ứng diễn ra sau đó sản sinh ra các loại oxy hoạt hóa (ROS) thì Nrf2 rời liên kết và trở thành hoạt hóa. Keap1 có các bộ phận thiol hoạt hóa liên kết ROS hay các loại arsenic ái lực electron như arsenic (III) monomethyl hóa và gây ra sự giải phóng Nrf2 để sau đó nó di chuyển qua tế bào chất tới nhân tế bào.[33] Nrf2 sau đó kích hoạt yếu tố phản ứng chống oxy hóa (ARE) cũng như yếu tố phản ứng ái lực electron (EpRE), cả hai đều góp phần vào việc gia tăng các protein chống oxy hóa.[34] Đáng chú ý trong các protein chống oxy hóa này là heme oxygenase 1 ([HO-1]), NAD(P)H-quinon oxidoreductase 1 (NQO1) và γ-glutamylcystein synthase (γGCS), làm việc trong sự liên kết để giảm các loại chất oxy hóa như Hydro peroxide để giảm ứng kích oxy hóa trong tế bào. Sự gia tăng trong γGCS gây ra sự sản xuất gia tăng của arsenit triglutathionin (As(SG)3), một sản phẩm cộng quan trọng được tiếp nhận bởi hoặc là protein MRP1 hoặc là protein MRP2, nhằm loại bỏ arsenic ra khỏi tế bào và đưa vào mật để bài tiết.[33] Cần lưu ý rằng sản phẩm cộng này cũng có thể bị phân hủy ngược trở lại thành arsenic vô cơ.
Đáng chú ý trong bài tiết arsenic là các bước methyl hóa nhiều lần diễn ra có thể làm tăng độc tính của arsen[35] do MMeAsIII là chất ức chế mạnh của glutathion peroxidase,[36] glutathion reductase, pyruvat dehydrogenase,[37] và thioredoxin reductase.[38]
Độc tính arsenic
[sửa | sửa mã nguồn]Arsenic là một nguyên nhân gây tử vong trên khắp thế giới; các vấn đề gắn liền với nó bao gồm các bệnh về tim, hô hấp, tiêu hóa, gan, thần kinh và thận.[2][24]
Arsenic can thiệp vào tuổi thọ tế bào bằng ức chế dị không gian một tổ hợp enzym trao đổi chất thiết yếu là pyruvat dehydrogenase (PDH), chất xúc tác sự oxy hóa pyruvat thành acetyl-CoA bởi NAD+. Với enzym bị ức chế, hệ thống năng lượng của tế bào bị đứt đoạn và gây ra chết rụng tế bào. Về mặt hóa sinh, arsenic ngăn cản sử dụng thiamin, gây ra hình ảnh lâm sàng tương tự như thiếu hụt thiamin. Ngộ độc arsenic có thể làm tăng mức lactat và dẫn tới nhiễm acid lactic.
Độc tính gen bao gồm ức chế sửa chữa DNA và methyl hóa DNA. Tác động gây ung thư của arsenic sinh ra từ ứng kích oxy hóa gây ra bởi arsenic. Độc tính cao của arsenic tự nhiên dẫn tới sự phát triển của một loạt các hợp chất arsenic như các vũ khí hóa học, chẳng hạn dimethylarsenic chloride. Một số trong các chất này đã từng được sử dụng như là tác nhân chiến tranh hóa học, đặc biệt trong Thế chiến I. Mối đe dọa này đã dẫn tới nhiều nghiên cứu về các thuốc giải độc và một kiến thức mở rộng về tương tác của các hợp chất arsenic với sinh vật. Một kết quả trong đó là sự phát triển của các thuốc giải độc, như dimercaprol. Nhiều loại thuốc giải độc tương tự như vậy khai thác ái lực của As(III) với các phối thể thiolat để chuyển hóa các hợp chất arsenic hữu cơ độc tính cao thành các chất ít độc hơn. Nói chung người ta cho rằng các arsenat gắn với các phần còn lại của cystein trong các protein.
Ngược lại, arsenic oxide lại được phê chuẩn và là một loại dược phẩm trị liệu hóa học có hiệu lực để điều trị ung thư bạch cầu tủy bào cấp tính (APL).[3]
Ngoài các dạng vô cơ, arsenic cũng tồn tại trong nhiều dạng hữu cơ trong môi trường. Arsenic vô cơ và các hợp chất của nó, khi đi vào chuỗi thức ăn, được trao đổi tích cực thành dạng ít độc hơn của arsenic thông qua quá trình methyl hóa. Ví dụ, Scopulariopsis brevicaulis, một loài nấm mốc sinh ra một lượng đáng kể trimethylarsin nếu arsenic vô cơ tồn tại. Hợp chất hữu cơ arsenobetain tìm thấy trong một số hải sản như cá và tảo, cũng như trong nấm ăn với hàm lượng lớn. Trung bình một người tiếp nhận khoảng 10-50 µg/ngày. Giá trị khoảng 1.000 µg không phải là bất thường sau khi tiêu thụ cá và nấm nhưng ở đây có rất ít nguy hiểm trong việc ăn cá do hợp chất arsenic trong cá là gần như không độc hại.[39]
Độc tính
[sửa | sửa mã nguồn]Độc tính của các hợp chất arsenic hóa trị 5
[sửa | sửa mã nguồn]Do cấu trúc và thuộc tính tương tự nên các chất trao đổi chất của arsenic hóa trị 5 có khả năng thay thế nhóm phosphat trong nhiều cách thức trao đổi chất.[40] Sự thay thế phosphat bằng arsenat được khởi đầu khi arsenat phản ứng với glucose và gluconat in vitro.[40] Phản ứng này sinh ra glucose-6-arsenat và 6-arsenogluconat, có tác động giống như glucose-6-phosphat và 6-phosphorgluconat.[40] Ở mức chất nền, trong thủy phân glucose, glucose-6-arsenat liên kết như là một chất nền cho glucose-6-phosphat dehydrogenase, và cũng ức chế hexokinase thông qua hồi tiếp âm.[40] Không giống như tầm quan trọng của phosphat trong thủy phân glucose, sự có mặt của arsenat hạn chế sự sản sinh ATP bằng cách tạo ra một sản phẩm anhydrid không ổn định, thông qua phản ứng với D-glyceraldehyd-3-phosphat.[40] Anhydrid 1-arsenato-3-phospho-D-glycerat sinh ra dễ dàng bị thủy phân do độ dài liên kết dài hơn của As-O so với P-O.[40] Ở mức ti thể, arsenat không ghép nối sự tổng hợp ATP bằng liên kết với ADP trong sự hiện diện của succinat, vì thế tạo ra một hợp chất không ổn định cuối cùng tạo ra sự sụt giảm của ATP tổng thể được sinh ra.[40] Ngược lại, các chất trao đổi chất arsenit (III) lại có tác động hạn chế tới sự sản xuất ATP trong hồng cầu.[40]
Độc tính của hợp chất arsenic hóa trị 3
[sửa | sửa mã nguồn]Các enzym và các thụ quan chứa thiol hoặc các nhóm chức sulfhydryl là mục tiêu của các chất trao đổi chất arsenit (III).[40] Các hợp chất chứa lưu huỳnh này thông thường là glutathion và amino acid cystein.[40] Các dẫn xuất arsenit nói chung có ái lực liên kết cao hơn so với các chất trao đổi chất arsenat.[40] Các liêt kết này ngăn trở hoạt động của một số cách thức trao đổi chất nhất định.[40] Chẳng hạn, pyruvat dehydrogenase (PDH) bị ức chế khi acid monomethylarsonơ (MMAIII) nhắm tới nhóm thiol của phụ nhân tử acid lipoic.[40] PDH là tiền chất của acetyl-CoA, vì thế ức chế PDH cuối cùng hạn chế sự sản xuất ATP trong chuỗi vận chuyển electron, cũng như sự sản xuất của các chất trung gian trong quá trình dị sinh glucose.[40]
Ứng kích oxy hóa
[sửa | sửa mã nguồn]Arsenic có thể gây ra ứng kích oxy hóa thông qua sự hình thành các loại oxy hoạt hóa (ROS) và các loại nitơ hoạt hóa (RNS).[27] Các loại oxy hoạt hóa được sinh ra bởi enzym NADPH oxidase, chuyển các electron từ NADPH sang oxy, tổng hợp một superoxide, là một gốc tự do hoạt hóa. Superoxide này có thể phản ứng để tạo thành hydro peroxid và các loại oxy hoạt hóa. Enzym NADPH oxidasecó khả năng sinh ra nhiều loại oxy hoạt hóa hơn khi có mặt arsenic do khối con p22phax chịu trách nhiệm vận chuyển electron bị điều chỉnh tăng bởi arsenic.[27] Các loại oxy hoạt hóa có khả năng ứng kích lưới nội bào, làm tăng lượng các tín hiệu phản ứng protein không gấp nếp.[27] Điều này dẫn tới viêm sưng, phát triển và phân chia tế bào và cuối cùng dẫn tới chết tế bào.[27] Một cơ chế khác trong đó các loại oxy hoạt hóa gây ra chết tế bào có thể là thông qua tái sắp xếp bộ xương tế bào có ảnh hưởng tới các protein có thể co lại.[27]
Các loại nitơ hoạt hóa sinh ra một khi các loại oxy hoạt hóa phá hủy ti thể.[27] Điều này dẫn đến sự hình thành của các loại nitơ hoạt hóa, tác nhân chịu trách nhiệm gây ra tổn thương AND trong ngộ độc arsenic.[27] Tổn thương ti thể được biết đến là nguyên nhân gây ra sự giải phóng các loại nitơ hoạt hóa, do phản ứng giữa các superoxide và nitơ monoxide (NO).[27] Nitơ monoxide (NO) là một phần của sự điều chỉnh tế bào; bao gồm trao đổi chất, phát triển, phân chia và chết tế bào.[27] Nitơ monoxide phản ứng với các loại oxy hoạt hóa để tạo thành peroxynitrit.[27] Trong các trường hợp phơi nhiễm arsenic mạn tính, các mức nitơ monoxide bị cạn kiệt, do các phản ứng của superoxide.[27] Enzym NO synthase (NOS) sử dụng L-arginin để tạo ra nitơ monoxide, nhưng enzyme này bị ức chế bởi các hợp chất arsenic (III) monomethyl hóa.[27]
Tổn thương DNA
[sửa | sửa mã nguồn]Arsenic được coi là nguyên nhân gây ra các biến đổi DNA như thể lệch bội, hình thành các vi nhân, dị thường nhiễm sắc thể, đột biến suy kiệt, trao đổi nhiễm sắc tử chị em và liên kết ngang của DNA với các protein.[41] Người ta đã chứng minh rằng arsenic không tương tác trực tiếp với DNA và nó được coi là một tác nhân gây đột biến kém, nhưng thay vì thế nó hỗ trợ tính gây đột biến các tác nhân gây ung thư khác.[42] Chẳng hạn, sự gia tăng hợp lực trong hoạt động gây đột biến của arsenic với tia cực tím đã được quan sát ở tế bào của người và các động vật có vú khác sau khi phơi nhiễm các tế bào đã xử lý bằng tia cực tím với arsenic.[43][44] Một loạt các quan sát thực nghiệm gợi ý rằng độc tính gen của arsenic chủ yếu liên kết với sự sản sinh các loại oxy hoạt hóa (ROS) trong biến đổi sinh học của nó.[45][46][47] Sự sản xuất ROS có thể sinh ra các sản phẩm cộng DNA, phá vỡ sợi DNA, liên kết ngang và các sai lệch nhiễm sắc thể.[48][49][50][51] Tổn thương oxy hóa bị gây ra bởi biến đổi của các nucleobase DNA, cụ thể là 8-oxoguanin (8-OHdG) dẫn tới các đột biến G:C thành T:A.[52] Arsenic vô cơ cũng có thể gây ra phá vỡ sợi DNA ngay cả khi ở các nồng độ thấp.[53]
Ức chế sửa chữa DNA
[sửa | sửa mã nguồn]Ức chế các quá trình sửa chữa DNA được coi là một trong các cơ chế chính của độc tính gen của arsenic vô cơ. Sửa chữa cắt xén nucleotide (NER) và sửa chữa cắt xén base (BER) là các quá trình kéo theo trong sửa chữa tổn thương base DNA gây ra bởi ROS sau phơi nhiễm arsenic. Cụ thể, cơ chế NER là cách thức chủ yếu trong sửa chữa các biến dạng lớn trong chuỗi xoắn kép DNA, trong khi cơ chế BER là chủ yếu trong sửa chữa các đứt gãy sợi đơn gây ra bởi ROS,[54][55][56][57] nhưng arsenic vô cơ cũng có thể ngăn chặn cơ chế BER.[58][59][59][60]
Cơ chế suy thoái thần kinh
[sửa | sửa mã nguồn]Arsenic là rất bất lợi cho hệ miễn dịch bẩm sinh và thích ứng của cơ thể.[61] Khi một lượng protein không gấp nếp và gấp nếp sai trong ứng kích lưới nội bào là thái quá thì phản ứng protein không gấp nếp (UPR) được hoạt hóa để làm tăng độ hoạt động của một vài thụ quan chịu trách nhiệm cho sự phục hồi của cân bằng nội môi.[61] Inositol-requiring enzyme-1 (IRE1) và protein kinase RNA-like endoplasmic reticulum kinase (PERK) là 2 thụ quan hạn chế tốc độ dịch mã.[61] Ngược lại, các protein không gấp nếp được hiệu chỉnh bởi sự sản xuất các chaperon, gây ra bởi nhân tử phiên mã hoạt hóa 6 (ATF6).[61] Nếu số lượng protein mắc lỗi tăng lên, cơ chế tiếp theo được kích hoạt để khởi động chết rụng tế bào.[61] Có chứng cứ chỉ ra là arsenic làm tăng hoạt tính của các cảm biến protein này.[61]
Rối loạn chức năng miễn dịch
[sửa | sửa mã nguồn]Phơi nhiễm arsenic ở trẻ em làm biến đổi tỷ lệ tế bào T phụ trợ (CD4) trên tế bào T độc tính tế bào (CD8), là yếu tố chịu trách nhiệm cho sụt giảm miễn dịch.[62] Ngoài ra, arsenic cũng làm tăng số lượng các phân tử viêm nhiễm được tiết ra thông qua các đại thực bào.[62] Lượng thái quá các bạch cầu hạt và bạch cầu đơn nhân dẫn tới trạng thái viêm nhiễm mạn tính, và có thể dẫn đến phát triển ung thư.[62]
Xử lý ngộ độc arsenic
[sửa | sửa mã nguồn]Có ba phân tử có vai trò như các tác nhân càng hóa liên kết với arsenic. Ba chất này là Dimercaprol (BAL, British Anti-Lewisite), succimer (DMSA) và Unithiol (DMPS).[63]
Khi các tác nhân này càng hóa arsenic vô cơ, nó được chuyển thành dạng hữu cơ của arsenic do nó được liên kết với tác nhân càng hóa hữu cơ. Các nguyên tử lưu huỳnh của các nhóm thiol là khu vực tương tác với arsenic. Điều này là do các nhóm thiol có ái lực hạt nhân trong khi các nguyên tử arsenic có ái lực electron. Một khi đã liên kết với tác nhân tạo càng thì các phân tử có thể được bài tiết, và vì thế các nguyên tử arsenic vô cơ tự do bị loại bỏ khỏi cơ thể.
Các tác nhân tạo càng khác cũng có thể được sử dụng, nhưng có thể gây ra các tác dụng phụ nhiều hơn so với dimercaprol, DMSA và DMPS. DMPS và DMSA cũng có chỉ số trị liệu (TI) cao hơn so với BAL.[63]
Các dược phẩm này có hiệu lực với ngộ độc arsenic cấp tính, nghĩa là với các hiệu ứng tức thời của ngộ độc arsenic. Chẳng hạn đau đầu, nôn mửa hay đổ mồ hôi là những ví dụ chung về hiệu ứng tức thời. Để so sánh thì các hiệu ứng ngộ độc mạn tính phát sinh muộn hơn và xảy ra bất ngờ, như các tổn thương cơ quan. Thông thường thì là quá muộn để ngăn chặn chúng một khi chúng xuất hiện. Vì thế, hành động nên được thực hiện càng sớm càng tốt khi các hiệu ứng ngộ độc cấp tính xuất hiện.[64]
Xem thêm
[sửa | sửa mã nguồn]- Hợp chất arsenic
- Sinh vật chịu điều kiện cực đoan
- Địa vi sinh học
- Các kiểu giả thuyết của hóa sinh học
- Hóa học arsenic hữu cơ
Tham khảo
[sửa | sửa mã nguồn]- ^ Pearce, Fred (2006). When the Rivers Run Dry: Journeys Into the Heart of the World's Water Crisis. Toronto: Key Porter. ISBN 978-1-55263-741-8.
- ^ a b c d Elke Dopp, Andrew D. Kligerman & Roland A. Diaz-Bone, 2010. Organoarsenicals. Uptake, Metabolism, and Toxicity, Royal Society of Chemistry. ISBN 978-1-84973-082-2. doi:10.1039/9781849730822-00231
- ^ a b Wilcox, Dean E. (2013). “Chapter 15. Arsenic. Can This Toxic Metalloid Sustain Life?”. Trong Astrid Sigel, Helmut Sigel & Roland K. O. Sigel (biên tập). Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. 13. Springer. tr. 475–498. doi:10.1007/978-94-007-7500-8_15.
- ^ Arsenocholine - Cấu trúc và Dữ liệu
- ^ Francesconi, Kevin A.; Edmonds, John S.; Stick, Robert V. (1992). “Arsenic compounds from the kidney of the giant clam Tridacna maxima: Isolation and identification of an arsenic-containing nucleoside”. Journal of the Chemical Society, Perkin Transactions 1 (11): 1349. doi:10.1039/P19920001349.
- ^ Bentley, Ronald; Chasteen, TG (2002). “Microbial Methylation of Metalloids: Arsenic, Antimony, and Bismuth”. Microbiology and Molecular Biology Reviews. 66 (2): 250–271. doi:10.1128/MMBR.66.2.250-271.2002. PMC 120786. PMID 12040126.
- ^ Cullen, William R; Reimer, Kenneth J. (1989). “Arsenic speciation in the environment”. Chemical Reviews. 89 (4): 713–764. doi:10.1021/cr00094a002.
- ^ Ronald Bentley & Thomas G. Chasteen (2002). “Microbial Methylation of Metalloids: Arsenic, Antimony, and Bismuth”. Microbiology and Molecular Biology Reviews. 66 (2): 250–271. doi:10.1128/MMBR.66.2.250-271.2002. PMC 120786. PMID 12040126.
- ^ Cullen, William R.; Reimer, Kenneth J. (1989). “Arsenic speciation in the environment”. Chemical Reviews. 89: 713–764. doi:10.1021/cr00094a002.
- ^ Oremland, Ronald S.; Saltikov, Chad W.; Wolfe-Simon, Felisa; Stolz, John F. (2009). “Arsenic in the Evolution of Earth and Extraterrestrial Ecosystems”. Geomicrobiology Journal. 26 (7): 522–536. doi:10.1080/01490450903102525.
- ^ Erb, T. J.; Kiefer, P.; Hattendorf, B.; Gunther, D.; Vorholt, J. A. (2012). “GFAJ-1 is an Arsenate-Resistant, Phosphate-Dependent Organism”. Science. 337 (6093): 467–70. Bibcode:2012Sci...337..467E. doi:10.1126/science.1218455. PMID 22773139.
- ^ Reaves, M. L.; Sinha, S.; Rabinowitz, J. D.; Kruglyak, L.; Redfield, R. J. (2012). “Absence of Detectable Arsenate in DNA from Arsenate-Grown GFAJ-1 Cells”. Science. 337 (6093): 470–3. arXiv:1201.6643. Bibcode:2012Sci...337..470R. doi:10.1126/science.1219861. PMC 3845625. PMID 22773140.
- ^ Westheimer, F.H. (ngày 6 tháng 6 năm 1987). “Why nature chose phosphates” (PDF). Science. 235 (4793): 1173–1178 (see pp. 1175–1176). Bibcode:1987Sci...235.1173W. doi:10.1126/science.2434996. Bản gốc (PDF) lưu trữ ngày 16 tháng 6 năm 2011.
- ^ Nordstrom D. K. (2002). “Worldwide occurrences of arsenic in ground water”. Science. 296 (5576): 2143–2145. doi:10.1126/science.1072375.
- ^ Hileman, B (ngày 9 tháng 4 năm 2007). “Arsenic in Chicken Production”. Chemical and Engineering News. tr. 34–35.
- ^ Bottemiller, Helena (ngày 26 tháng 9 năm 2009). “Bill Introduced to Ban Arsenic Antibiotics in Feed”. Food Safety News. Truy cập ngày 10 tháng 1 năm 2011.
- ^ a b Sakurai T (2003). “Biomethylation of Arsenic is Essentially Detoxicating Event”. Journal of Health Science. 49 (3): 171–178. doi:10.1248/jhs.49.171. Truy cập ngày 10 tháng 1 năm 2011.
- ^ Jun Zhu; Zhu Chen; Valérie Lallemand-Breitenbach; Hugues de Thé (2002). “How Acute Promyelocytic Leukaemia Revived Arsenic”. Nature Reviews Cancer. 2 (9): 705–714. doi:10.1038/nrc887. PMID 12209159. Bản gốc lưu trữ ngày 19 tháng 10 năm 2017. Truy cập ngày 27 tháng 9 năm 2013.
- ^ a b Gibaud, Stéphane; Jaouen, Gérard (2010). “Arsenic - based drugs: from Fowler's solution to modern anticancer chemotherapy”. Topics in Organometallic Chemistry. Topics in Organometallic Chemistry. 32: 1–20. doi:10.1007/978-3-642-13185-1_1. ISBN 978-3-642-13184-4.
- ^ Elschenbroich C., 2006. "Organometallics". Wiley-VCH: Weinheim. ISBN 978-3-527-29390-2
- ^ a b Park, Woo H. Park; Jae G. Seol; Eun S. Kim; Jung M. Hyun; Chul W. Jung; Chung C. Lee; Byoung K. Kim; Young Y. Lee (ngày 6 tháng 6 năm 2000). “Arsenic Trioxide-mediated Growth Inhibition in MC/CAR Myeloma Cells via Cell Cycle Arrest in Association with Induction of Cyclin-dependent Kinase Inhibitor, p21, and Apoptosis”. Cancer Research. 60 (3065): 3065–71. PMID 10850458. Truy cập ngày 15 tháng 12 năm 2010.
- ^ Lunghi, Paolo Lunghi; Antonio Costanzo; Massimo Levrero; Antonio Bonati (ngày 15 tháng 7 năm 2004). “Treatment with arsenic trioxide (ATO) and MEK1 inhibitor activates the p73-p53AIP1 apoptotic pathway in leukemia cells”. Blood. 104 (2): 519–525. doi:10.1182/blood-2003-08-2743. PMID 15031205. Truy cập ngày 15 tháng 12 năm 2010.
- ^ “Arsenic in Drinking Water - Review article” (PDF). IARC Monographs - World Health Organization. 84: 133–135. Truy cập ngày 10 tháng 1 năm 2011.
- ^ a b c “Arsenic in Drinking Water - Review article” (PDF). IARC Monographs - World Health Organization. 84. Truy cập ngày 10 tháng 1 năm 2011.
- ^ a b c d e f g h i j Kumagai, Yoshito; Sumi, Daigo (2007). “Arsenic: Signal Transduction, Transcription Factor, and Biotransformation Involved in Cellular Response and Toxicity”. Annual Review of Pharmacology and Toxicology. 47: 243–62. doi:10.1146/annurev.pharmtox.47.120505.105144. PMID 17002598.
- ^ a b Vahter, Marie (2002). “Mechanisms of arsenic biotransformation”. Toxicology. 181–182: 211–7. doi:10.1016/S0300-483X(02)00285-8. PMID 12505313.
- ^ a b c d e f g h i j k l m Hunt, Katherine M.; Srivastava, Ritesh K.; Elmets, Craig A.; Athar, Mohammad (2014). “The mechanistic basis of arsenicosis: Pathogenesis of skin cancer”. Cancer Letters. 354 (2): 211–9. doi:10.1016/j.canlet.2014.08.016. PMID 25173797.
- ^ Petrick, Jay S.; Ayala-Fierro, Felix; Cullen, William R.; Carter, Dean E.; Vasken Aposhian, H. (2000). “Monomethylarsonous Acid (MMAIII) is More Toxic Than Arsenite in Chang Human Hepatocytes”. Toxicology and Applied Pharmacology. 163 (2): 203–7. doi:10.1006/taap.1999.8872. PMID 10698679.
- ^ “Arsenic in Drinking Water - Review article” (PDF). IARC Monographs - World Health Organization. 84: 138. Truy cập ngày 10 tháng 1 năm 2011.
- ^ Ajees, A.A.; và đồng nghiệp (ngày 10 tháng 7 năm 2012). “Structure of an As(III) S-Adenosylmethionine Methyltransferase: insights into the Mechanism of Arsenic Biotransformation”. Biochemistry. 51: 5476–5485. doi:10.1021/bi3004632.
- ^ Kumagai, Yoshito; Sumi, Daigo Sumi (2007). “Arsenic: Signal Transduction, Transcription Factor, and Biotransformation Involved in Cellular Response and Toxicity”. Annual Review of Pharmacology and Toxicology. 47: 243–62. doi:10.1146/annurev.pharmtox.47.120505.105144. PMID 17002598.
- ^ Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J. D.; Yamamoto, M (1999). “Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain”. Genes Dev. 13 (1): 76–86. doi:10.1101/gad.13.1.76. PMC 316370. PMID 9887101.
- ^ a b Kumagai, Yoshito; Sumi, Daigo Sumi (2007). “Arsenic: Signal Transduction, Transcription Factor, and Biotransformation Involved in Cellular Response and Toxicity”. Annual Review of Pharmacology and Toxicology. 47: 243–62. doi:10.1146/annurev.pharmtox.47.120505.105144. PMID 17002598.
- ^ Pi, J; Waalkes, MP; Kumagai, Y; Reece, JM; Qu, W (2003). “Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide”. Exp. Cell Res. 290 (2): 234–45. doi:10.1016/s0014-4827(03)00341-0. PMID 14567983.
- ^ Stýblo, M.; Drobná, Z.; Jaspers, I.; Lin, S.; Thomas, D. J. (2002). “The role of biomethylation in toxicity and carcinogenicity of arsenic: A research update”. Environmental Health Perspectives. 110 (Suppl 5): 767–771. doi:10.1289/ehp2110s5767. PMC 1241242. PMID 12426129.
- ^ Chouchane, S.; Snow, E. T.; Snow, E. T. “In vitro effect of arsenical compounds on glutathione-related enzymes”. Chem. Res. Toxicol. 14 (5): 517–22. PMID 11368549.
- ^ Petrick, Jay S.; Jagadish, Bhumasamudram; Mash, Eugene A.; Aposhian, H. Vasken (2001). “Monomethylarsonous Acid (MMAIII) and Arsenite: LD50in Hamsters and in Vitro Inhibition of Pyruvate Dehydrogenase”. Chemical Research in Toxicology. 14 (6): 651–656. doi:10.1021/tx000264z. PMID 11409934.
- ^ Lin, Lin S.; Thomas, D. J.; Cullen, W. R.; Wang, C.; Styblo, M.; Del Razo, L. M. (2001). “Arsenicals inhibit thioredoxin reductase in cultured rat hepatocytes”. Chem. Res. Toxicol. 14 (3): 305–11. doi:10.1021/tx0001878. PMID 11258980.
- ^ Cullen, William R; Reimer, Kenneth J. (1989). “Arsenic speciation in the environment”. Chemical Reviews. 89 (4): 713–764. doi:10.1021/cr00094a002.
- ^ a b c d e f g h i j k l m n Hughes, Michael F (2002). “Arsenic toxicity and potential mechanisms of action”. Toxicology Letters. 133 (1): 1–16. doi:10.1016/S0378-4274(02)00084-X. PMID 12076506.
- ^ Rossman, T.G (2003). “Mechanism of arsenic carcinogenesis: An integrated approach”. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 533: 37–65. doi:10.1016/j.mrfmmm.2003.07.009.
- ^ Pierce, B.L; Kibriya, M.G (2012). “Genome-wide association study identifies chromosome 10q24.32 variants associated with arsenic metabolism and toxicity phenotypes in Bangladesh”. PLoS Genetics. 8: e1002522. doi:10.1371/journal.pgen.1002522.
- ^ Li, J.H; Rossman, T.G (1991). “Comutagenesis of sodium arsenite with ultraviolet radiation in Chinese hamster V79 cells”. Biology of Metals. 4: 197–200. doi:10.1007/BF01141180.
- ^ Lee, T.C; Oshimura, M (1985). “Comparison of arsenic-induced cell transformation, cytotoxicity, mutation and cytogenetic effects in Syrian hamster embryo cells in culture”. Carcinogenesis. 6: 1421–1426. doi:10.1093/carcin/6.10.1421.
- ^ Kessel, M; Liu, S.X (2002). “Arsenic induces oxidative DNA damage in mammalian cells”. Molecular and Cellular Biochemistry. 234/235: 234–235:301–308. doi:10.1023/A:1015927406142.
- ^ Nesnow, S; Roop, B.C (2002). “DNA damage induced by methylated trivalent arsenicals is mediated by reactive oxygen species”. Chemical Research in Toxicology. 15: 1627–1634. doi:10.1021/tx025598y.
- ^ Jomova, K; Jenisova, Z (2011). “Arsenic: Toxicity, oxidative stress and human disease”. Journal of Applied Toxicology. 31: 95–107. doi:10.1002/jat.1649.
- ^ Kitchin, K.T; Wallace, K (2008). “Evidence against the nuclear in situ binding of arsenicals—Oxidative stress theory of arsenic carcinogenesis”. 232: 252–257. doi:10.1016/j.taap.2008.06.02. Chú thích journal cần
|journal=(trợ giúp) - ^ Bau, D.T; Wang, T.S (2002). “Oxidative DNA adducts and DNA-protein cross-links are the major DNA lesions induced by arsenite”. Environmental Health Perspectives. 110: 753–756. doi:10.1289/ehp.02110s5753. PMC 1241239. PMID 12426126.
- ^ Hwang, E.S; Kim, G.H (2007). “Biomarkers for oxidative stress status of DNA, lipids, and proteins in vitro and in vivo cancer research”. Toxicology. 229: 1–10. doi:10.1016/j.tox.2006.10.013.
- ^ Liu, Su X. (tháng 12 năm 2000). “Induction of oxyradicals by arsenic: Implication for mechanism of genotoxicity”. Proceedings of the National Academy of Sciences of the United States of America. Truy cập ngày 4 tháng 4 năm 2013.
- ^ Grollman, A.P; Moriya, M (1993). “Mutagenesis by 8-oxoguanine: An enemy within”. Trends in Genetics. 9: 246–249. doi:10.1016/0168-9525(93)90089-Z.
- ^ Martinez, V.D; Vucic, E.A (2011). “Arsenic biotransformation as a cancer promoting factor by inducing DNA damage and disruption of repair mechanisms”. Molecular Biology International. 2011: 1–11. doi:10.4061/2011/718974.
- ^ Lai, Y; Zhao, W (2011). “Role of DNA polymerase beta in the genotoxicity of arsenic”. Environmental and Molecular Mutagenesis. 52: 460–468. doi:10.1002/em.20643.
- ^ Hartwig, A; Groblinghoff, U.D (1997). “Interaction of arsenic(III) with nucleotide excision repair in UV-irradiated human fibroblasts”. Carcinogenesis. 18: 399–405. doi:10.1093/carcin/18.2.399. PMID 9054635.
- ^ Curnow, A; Salter, L (2001). “A preliminary investigation of the effects of arsenate on irradiation-induced DNA damage in cultured human lung fibroblasts”. Journal of Toxicology and Environmental Health, Part A. 63: 605–616. doi:10.1080/152873901316857789.
- ^ Schwerdtle, T; Walter, I (2003). “Induction of oxidative DNA damage by arsenite and its trivalent and pentavalent methylated metabolites in cultured human cells and isolated DNA”. Carcinogenesis. 24: 967–974. doi:10.1093/carcin/bgg018.
- ^ Lai, Y; Zhao, W (2011). “Role of DNA polymerase beta in the genotoxicity of arsenic”. Environmental and Molecular Mutagenesis. 52: 460–468. doi:10.1002/em.20643.
- ^ a b Ebert, F; Weiss, A (2011). “Arsenicals affect base excision repair by several mechanisms”. Mutat Res. 715: 32–41. doi:10.1016/j.mrfmmm.2011.07.004. PMID 21782832.
- ^ Sykora, P; Snow, E.T (2008). “Modulation of DNA polymerase beta-dependent base excision repair in cultured human cells after low dose exposure to arsenite”. Toxicology and Applied Pharmacology. 228: 385–394. doi:10.1016/j.taap.2007.12.019.
- ^ a b c d e f Hunt, K. M; Srivastava, R. K; Elmets, C. A; Athar, M. (2014). “The mechanistic basis of arsenicosis: Pathogenesis of skin cancer”. Cancer Letters. 354: 211–219. doi:10.1016/j.canlet.2014.08.016. PMID 25173797.
- ^ a b c Vega L. Environmental Health Risks. Nova Science Publishers. Tr. 157-159. ISBN 978-1-60741-781-1
- ^ a b Kosnett, M. J. (2013). “The Role of Chelation in the Treatment of arsenic and Mercury Poisoning”. Journal of medical toxicology. 4: 347–357. doi:10.1007/s13181-013-0344-5.
- ^ “Acute & Chronic Poisoning Affects”. medtox. Bản gốc lưu trữ ngày 16 tháng 8 năm 2015. Truy cập ngày 30 tháng 3 năm 2015.